If you want to model protein interactions, metabolic pathways, and cell signaling simultaneously — a multi-agent AI workflow is the most powerful approach available today. This guide walks through exactly how to build one, what each agent does, and why this architecture outperforms every traditional single-model approach in computational biology.
The Problem With Traditional Systems Biology Pipelines
Modern biology is inherently multi-layered. A single cancer cell at any moment is running thousands of concurrent processes — gene expression cascades firing, proteins binding and dissociating, metabolic flux shifting in response to nutrient availability, and signaling cascades propagating through receptors to the nucleus. The challenge? Traditional computational tools treat each of these as a separate problem.
Modern systems biology often suffers from fragmented data silos where regulatory networks, metabolic pathways, and signaling cascades are analyzed in isolation. A researcher might use one tool for gene regulatory networks, another for protein-protein interaction prediction, and a third for metabolic flux analysis — and then manually try to piece together what it all means.
This is where a multi-agent AI workflow fundamentally changes the game.
What Is a Multi-Agent AI Workflow? (Definition + Expansion)
A multi-agent AI workflow is an architecture where multiple specialized AI models — each an “agent” — handle distinct subtasks in parallel, then hand off their outputs to a coordinating agent that synthesizes the results into a unified answer.
In the context of biological network modeling, each agent is trained or prompted to handle a specific domain of computational biology. Rather than forcing a single language model to be an expert in gene regulation and protein biochemistry and metabolic engineering and systems pharmacology at the same time, you divide and conquer.
This multi-agent approach addresses the technical reality of fragmented analysis by using an LLM-based Principal Investigator agent to synthesize disparate outputs, ensuring that individual agent findings are connected into a broader, biologically plausible narrative.
Think of it as assembling a virtual research team: a genomicist, a structural biologist, a metabolic engineer, and a cell biologist — all working in parallel, reporting to a PI who writes the final integrated report.
Why Systems Biology Demands a Multi-Agent Approach
The Scale Problem
In a complete systems biology pipeline, researchers need to generate synthetic biological data, analyze gene regulatory structure, predict protein-protein interactions, optimize metabolic pathway activity, and simulate a dynamic cell signaling cascade — all in one unified workflow. No single model handles all of these tasks with equal depth.
The Integration Problem
Multi-agent structures function as a virtual interdisciplinary team, integrating expertise across data science, biology, and clinical research into a cohesive analytical unit — reducing communication overhead, accelerating analysis, and enabling individual users to achieve team-level productivity.
The Reproducibility Problem
When different tools from different labs are stitched together manually, reproducing results becomes nearly impossible. A unified multi-agent AI workflow running in a single environment (like a Google Colab notebook) provides end-to-end reproducibility with version-controlled logic at every stage.
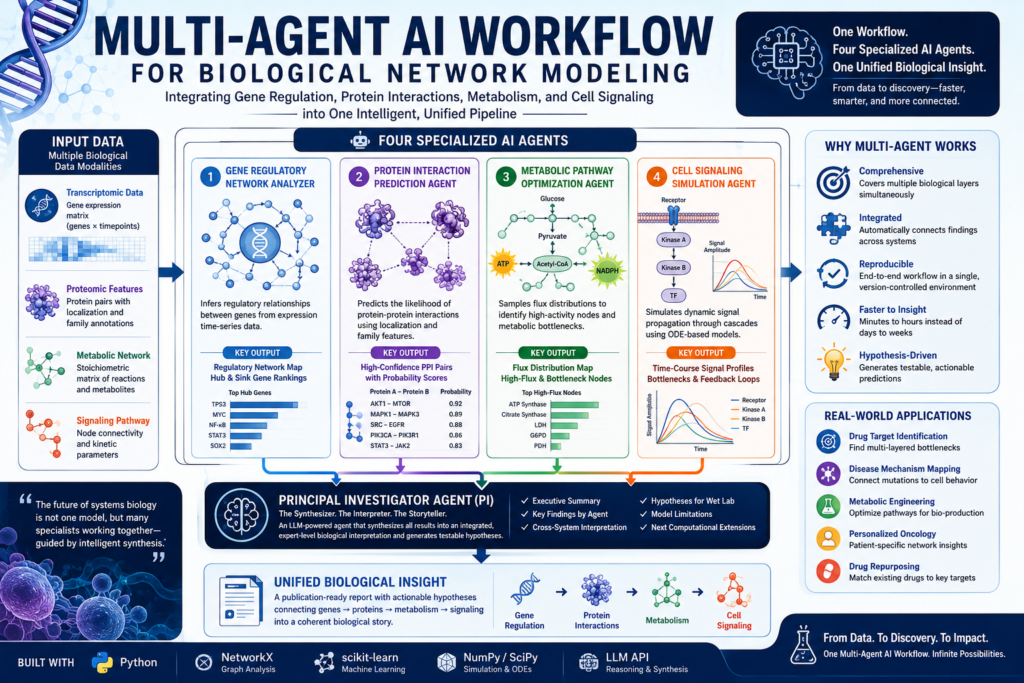
The Four Specialized Agents: What Each One Does
The core of a multi-agent AI workflow for biological network modeling rests on four specialized agents. Here is how each one works:
Agent 1 — Gene Regulatory Network (GRN) Analyzer
What it does: The GRN agent takes simulated or real gene expression time-series data and infers regulatory relationships between genes.
The Gene Regulatory Network Agent uses correlation-based association inference and NetworkX to identify hub and sink genes from simulated expression trajectories.
Hub genes are those with many regulatory connections — often master transcription factors like TP53, MYC, or NF-κB. Sink genes receive many regulatory signals but send few, making them candidate end-effectors of gene programs. By mapping these relationships, the GRN agent builds a directed graph that shows who is controlling whom at the transcriptional level.
Key output: A regulatory adjacency matrix and a ranked list of hub and sink gene candidates.
Agent 2 — Protein Interaction Prediction Agent
What it does: This agent takes a list of candidate protein pairs and scores the likelihood that they physically interact.
The Protein Interaction Prediction Agent employs Logistic Regression via Scikit-learn to rank candidate protein pairs based on shared localization and family features.
Proteins that share subcellular localization (e.g., both in the nucleus) and belong to related protein families (e.g., both kinases) are far more likely to interact than random pairs. The agent uses these structural and functional features to generate an interaction probability score for every candidate pair — essentially building a protein-protein interaction (PPI) network de novo from feature data.
Key output: A ranked list of high-confidence protein-protein interaction pairs with probability scores.
Agent 3 — Metabolic Pathway Optimization Agent
What it does: This agent models the metabolic network as a system of flux-balance equations and identifies which enzyme activities maximize a target output — such as ATP production, NADPH generation, or biosynthesis of a key metabolite.
By automating the integration of correlation-based association inference and randomized flux searches, the workflow provides a scalable framework for complex hypothesis generation that exceeds the capacity of manual data correlation.
The metabolic agent uses randomized flux sampling to explore the feasible solution space. Rather than finding a single “optimal” flux state (which may not be biologically realistic), it samples many plausible states and reports which metabolic nodes show the most consistent high activity — flagging them as likely bottlenecks or control points.
Key output: A metabolic flux distribution map with highlighted high-variance and high-flux nodes.
Agent 4 — Cell Signaling Simulation Agent
What it does: This agent models the dynamic propagation of a signaling cascade — for example, from receptor activation at the cell membrane through kinase phosphorylation chains to transcription factor activation in the nucleus.
The agent uses ordinary differential equations (ODEs) to simulate how signal amplitude changes over time through each node in the cascade. It captures phenomena like signal amplification, feedback inhibition, and signal decay — things that static network maps completely miss.
Key output: Time-course plots of signal amplitude at each node, with identification of cascade bottlenecks and feedback loops.
The Principal Investigator Agent: Where It All Comes Together
After the four specialized agents complete their analyses, the multi-agent AI workflow passes all results to a coordinating agent — the Principal Investigator (PI) agent — powered by a large language model.
The PI agent synthesizes the outputs of all specialized agents into a single expert-style biological interpretation that connects regulation, interaction networks, metabolism, and signaling into a broader scientific story.
The PI agent is prompted with a structured report format that includes:
- Executive Summary — the one-paragraph “so what” for the entire analysis
- Key Findings by Agent — what each specialist found, in plain scientific language
- Cross-System Biological Interpretation — how the gene regulation findings connect to the protein interactions, and how those interact with the metabolic flux state and the signaling dynamics
- Hypotheses Worth Testing in the Wet Lab — actionable, falsifiable predictions for experimental validation
- Model Limitations — honest acknowledgment of what the synthetic data or algorithmic assumptions might miss
- Next Computational Extensions — suggestions for deepening the analysis
This final synthesis is what makes the multi-agent AI workflow genuinely more powerful than the sum of its parts. No human researcher would have the bandwidth to simultaneously hold the GRN topology, the PPI network, the flux distribution, and the signaling dynamics in working memory and reason across all four. The PI agent does exactly this.
Multi-Agent AI Workflow vs. Traditional Computational Biology: A Comparison
| Feature | Traditional Pipeline | Multi-Agent AI Workflow |
|---|---|---|
| Analysis scope | Single biological layer at a time | All four layers simultaneously |
| Integration | Manual, researcher-dependent | Automated by PI agent |
| Reproducibility | Tool-dependent, often poor | End-to-end in one environment |
| Hypothesis generation | Researcher-driven post-analysis | Built into the synthesis step |
| Scalability | Requires separate experts per domain | Scales with LLM capabilities |
| Time to insight | Days to weeks | Minutes to hours |
| Wet lab connection | Rarely explicit | Testable hypotheses as output |
| Accessibility | Requires deep domain expertise | Accessible via natural language prompts |
The contrast is stark. A traditional pipeline might take a team of four domain specialists several weeks to complete what a well-designed multi-agent AI workflow can accomplish in a single Colab session.
How to Build a Multi-Agent AI Workflow for Biological Network Modeling
Here is a practical step-by-step overview of the architecture:
Step 1: Generate or Load Your Biological Data
The pipeline begins with synthetic or real data across four data modalities:
- Transcriptomic data — gene expression matrix (genes × timepoints)
- Proteomic feature data — protein pairs with localization and family annotations
- Metabolic network data — stoichiometric matrix of reactions and metabolites
- Signaling pathway data — node connectivity and kinetic parameters
In a tutorial or proof-of-concept setting, all four data types can be synthetically generated with numpy and scipy — creating realistic-looking datasets without requiring access to proprietary databases.
Step 2: Define and Deploy Each Specialized Agent
Each agent is implemented as a Python function (or class) that:
- Accepts its specific data modality as input
- Runs its computational analysis (NetworkX graph analysis, scikit-learn classification, flux sampling, ODE integration)
- Returns a structured summary — either a dictionary, a DataFrame, or a natural-language description
In a multi-agent AI workflow powered by an LLM backbone, each agent function can also be wrapped in an LLM call, allowing the model to interpret intermediate results and flag anomalies before passing them downstream.
Step 3: Aggregate Outputs Into the PI Agent Context
After all four agents complete their runs, their outputs are concatenated into a single prompt context window for the PI agent. The prompt includes:
- The raw numerical findings (top hub genes, top interacting protein pairs, high-flux metabolic nodes, signaling cascade amplitudes)
- Instructions for the report structure
- A constraint against data fabrication — the model must reason only from what was shown
Step 4: Run the PI Agent and Parse the Report
The PI agent is given the outputs of all four specialized agents and instructed to write a rigorous but readable report with sections covering an executive summary, key findings by agent, cross-system biological interpretation, hypotheses worth testing in a wet lab, model limitations, and next computational extensions.
The output is a structured scientific report, generated in seconds, that a domain expert could immediately use as a starting point for experimental planning.
Real-World Applications of This Architecture
The multi-agent AI workflow for biological network modeling isn’t just an academic exercise. Here are concrete use cases where this architecture delivers real value:
- Drug target identification — by connecting GRN hub genes to PPI network nodes to metabolic bottlenecks, the workflow can pinpoint proteins that are central across multiple biological layers — ideal drug targets
- Disease mechanism mapping — integrating transcriptomic data from patient samples with signaling simulation can reveal how a mutation propagates from gene regulation to cell behavior
- Metabolic engineering — in synthetic biology, the metabolic optimization agent can be used to design strains with enhanced production of target compounds
- Personalized oncology — tumor-specific transcriptomics can be fed into the GRN agent to identify patient-specific hub genes that drive cancer progression
- Drug repurposing — the cross-system synthesis step can identify existing drugs whose mechanisms of action align with the identified biological bottlenecks
Platforms like BioLab demonstrate that multi-agent AI systems can autonomously orchestrate the entire scientific discovery lifecycle, seamlessly integrating multi-round interactions between computational modeling and wet-lab experimentation to automate the full design-build-test-learn cycle.
Limitations and Honest Caveats
No technology is without limits. Here is what to keep in mind when deploying a multi-agent AI workflow for biological network modeling:
- Synthetic data ≠ real data — proof-of-concept pipelines using synthetic gene expression data may not generalize to messy, high-dimensional real-world omics datasets
- Correlation is not causation — the GRN agent infers associations, not causal regulatory relationships; validation requires perturbation experiments
- LLM hallucination risk — the PI agent must be explicitly constrained not to fabricate data; prompt engineering and system instructions are essential
- Computational cost — running ODE simulations and LLM synthesis in sequence can be resource-intensive at scale
- Biological ground truth — all hypotheses generated by the PI agent require wet lab validation before they can be trusted
While large language models provide some assistance with bioinformatics tasks, they often lack the nuanced guidance required for complex analyses and can be resource-intensive — motivating hybrid approaches that combine LLM reasoning with fine-tuned specialized models.
Frequently Asked Questions
What programming libraries are needed to build a multi-agent AI workflow for biology?
The core stack includes NetworkX (graph construction and analysis), scikit-learn (machine learning for protein interaction prediction), numpy/scipy (numerical simulation for ODE-based signaling models), and an LLM API (such as the OpenAI or Anthropic API) for the agent reasoning layer. All of these run comfortably in a Google Colab environment.
Can this multi-agent AI workflow work with real omics data?
Yes, with modification. The synthetic data generation functions in the tutorial version can be replaced with real data loaders for standard formats like CSV expression matrices, STRING database PPI files, BiGG metabolic models, or KEGG pathway data. The agent logic remains unchanged.
How many agents do I need?
Four specialized agents plus one PI (synthesizer) agent is a proven baseline. For more advanced applications, you can add agents for pathway enrichment analysis, drug-target docking scoring, or literature-based hypothesis retrieval — each feeding additional context into the PI agent’s synthesis step.
Is this approach reproducible?
Yes — because the entire multi-agent AI workflow runs in a single notebook with fixed random seeds and versioned dependencies, results can be reproduced by any researcher with access to the same environment and data.
What makes the Principal Investigator agent different from simply prompting a single LLM?
The PI agent has access to structured, pre-computed outputs from four domain-specific analytical agents. It is not being asked to reason from scratch about protein interactions or metabolic networks — it is being asked to synthesize pre-processed findings. This dramatically reduces hallucination risk and dramatically increases the scientific depth of the output.
The Future: Toward Fully Autonomous Systems Biology
The field has evolved rapidly from AI as an analytical tool to AI as an autonomous research collaborator capable of generating testable hypotheses, with systems reportedly accomplishing in one day what previously took six months of research work.
The multi-agent AI workflow described in this guide represents a crucial stepping stone toward that future. By assigning specialized agents to each layer of biological complexity and then connecting them through an intelligent synthesizer, we get something qualitatively new: a computational system that can reason across the full hierarchy of biological organization — from gene to protein to metabolite to cell behavior — and generate actionable scientific insights.
The next frontier will involve these workflows closing the loop: generating hypotheses, triggering automated wet lab experiments via robotic platforms, receiving experimental results, updating their models, and generating new hypotheses — all without human intervention between cycles.
We are not there yet. But the architecture to get there starts exactly with the multi-agent AI workflow design principles covered in this guide.
Key Takeaways
- A multi-agent AI workflow divides biological network modeling into four specialized agents — GRN analysis, protein interaction prediction, metabolic optimization, and cell signaling simulation
- A Principal Investigator agent synthesizes all four outputs into an integrated, publication-ready scientific report
- This architecture solves the fragmented data silo problem that has long plagued computational systems biology
- The full pipeline can be implemented in a single Google Colab environment using open-source Python libraries plus an LLM API
- Real-world applications span drug discovery, oncology, metabolic engineering, and personalized medicine
- Key limitations include synthetic data constraints, LLM hallucination risk, and the need for experimental validation of all generated hypotheses
Related Topics Worth Exploring
- Graph Neural Networks for Protein Interaction Prediction — moving beyond logistic regression to deep learning on molecular graphs
- Flux Balance Analysis (FBA) at Scale — integrating genome-scale metabolic models with transcriptomic constraints
- Causal Discovery in Gene Regulatory Networks — methods beyond correlation: GENIE3, ARACNE, and interventional data
- Agentic Scientific Discovery Platforms — BioLab, PromptBio, and the emerging ecosystem of autonomous research AI